Einführung
Das Virus des porcinen Reproduktions- und Respirationssyndroms (PRRSV) besitzt ein einsträngiges RNA-Genom, wodurch es sehr anfällig für genetische Mutationen wird. Dies macht aber auch jeden PRRSV-Stamm einzigartig, so dass die genetische Typisierung eine nützliche Methode zur Diagnose und Bekämpfung der Krankheit ist. Die Diagnose der genetischen Typisierung erfolgt durch DNA-Sequenzierung, also durch Bestimmung der Abfolge der Nukleotide in einer DNA-Kopie des viralen RNA-Genomfragments. Derzeit ist das für diese Zwecke am häufigsten verwendete Fragment ORF5, das Gen, welches das wichtigste Hüllglykoprotein kodiert, und zwar hauptsächlich deshalb, weil es eine große genetische Vielfalt aufweist.

Diagnostische DNA-Sequenzierung
Die Unterscheidung zwischen dem PRRSV Typ 1 (europäisch) und Typ 2 (amerikanisch) lässt sich von den meisten diagnostischen PCR-Tests leicht bewerkstelligen. Die Unterscheidung zwischen einzelnen Stämmen jedes der beiden Genotypen erfordert allerdings die DNA-Sequenzierung. Für diese Zwecke werden Körperflüssigkeiten oder Gewebe mit mäßigen bis großen Mengen PRRSV behandelt um RNA zu isolieren, die dann durch reverse Transkription in DNA kopiert wird. Anschließend wird das ORF5-Gen durch PCR amplifiziert und einem Verfahren zur DNA-Sequenzierung unterzogen. Dieser Vorgang läuft überwiegend automatisch ab und benötigt im Allgemeinen ein bis drei Tage. Die aus der Sequenzierung erhaltenen Rohdateien werden zur Analyse an das Diagnoselabor geschickt. Der Laborbefund beinhaltet normalerweise die Nukleotidsequenz des Stamms und seine Ähnlichkeit zu den üblichen Impfstämmen. Darüberhinaus bieten einige Labore auch einen Vergleich mit einer Standardliste von Wildtyp-Isolaten des PRRSV in Form eines Dendrogramms oder einen Vergleich mit einer PRRSV-Sequenzdatenbank eines Produktionssystems an.
Analyse von PRRSV-Sequenzen
Die Sequenzähnlichkeit oder -identität wird durch Alignieren von mindestens zwei Sequenzen mithilfe eines Computerprogramms bestimmt. Abb. 1 zeigt das Beispiel eines Alignments verschiedener ORF5-Sequenzen aus 5 verschiedenen Betrieben. Paarweise Vergleiche der prozentualen Identität sind in der Tabelle dargestellt. Die Identitäten reichen von 81,2 bis 99,8 %. Das Dendrogramm, das sich aus der phylogenetischen Analyse ergibt, weist eine Gruppierung der ähnlichen Sequenzen auf (Abb. 2). Eine Grundsatzfrage für Erzeuger und Veterinäre ist, ob die beobachteten genetischen Unterschiede zwischen den Sequenzen eine normale Abweichung eines einzelnen PRRSV-Stamms in einem Betrieb oder aber mehrere verschiedene Stämme darstellen, die in einem Betrieb vorhanden sind.

Abbildung 1: Abschnitt des Alignments von ORF5-Sequenzen von PRRSV-Stämmen aus 5 verschiedenen Betrieben. Aus den Betrieben 1, 4 und 5 erhielt man mehrere Sequenzen. Die Punkte in den Sequenzen entsprechen Stellen, die mit dem Lelystad Referenzstamm PRRSV Typ 1 identisch sind.
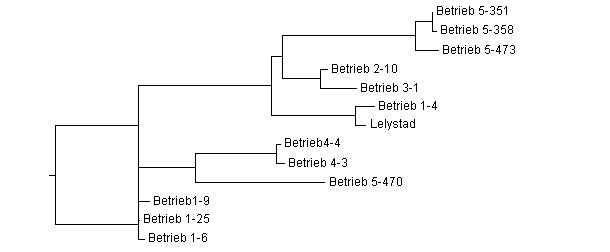
Abbildung 2: Dendrogramm von ORF5-Sequenzen aus 5 verschiedenen Betrieben. Interpretationsbeispiel: In Betrieb 1 gibt es zwei Stämme, die nicht miteinander verwandt sind. Drei Sequenzen sind >99% identisch, wohingegen die vierte nur zu ~83 % verwandt ist. Stattdessen ist sie mit dem Lelystad-Virus verwandt. Die Stämme aus Betrieb 2 und Betrieb 3 sind eng miteinander verwandt (98,2 % identisch). Zwei Stämme aus Betrieb 4 sind eng miteinander verwandt (99,5 % identisch). In Betrieb 5 gibt es zwei Stämme, die nicht miteinander verwandt sind. Drei Stämme sind >98 % identisch und ca. 81 % identisch mit dem vierten Stamm.
Tabelle: Paarweise prozentuale Gleichheit zwischen allen alignierten ORF5-Sequenzen einer Serie von Proben des PRRSV Typ 1.
| 1-4 | 1-6 | 1-9 | 1-25 | 2-10 | 3-1 | 4-4 | 4-3 | 5-351 | 5-358 | 5-470 | 5-473 | Lelystad | |
| *** | 83.5 | 83.3 | 83.7 | 93.2 | 92.6 | 86.1 | 86.0 | 88.1 | 88.0 | 83.2 | 88.3 | 98.7 | 1-4 |
| *** | 99.2 | 99.7 | 84.2 | 83.0 | 86.0 | 86.0 | 81.4 | 81.2 | 86.3 | 81.4 | 82.1 | 1-6 | |
| *** | 99.5 | 84.5 | 83.3 | 85.6 | 85.6 | 81.4 | 81.2 | 86.5 | 81.4 | 81.9 | 1-9 | ||
| *** | 84.5 | 83.3 | 86.0 | 86.0 | 81.7 | 81.5 | 86.8 | 81.7 | 82.2 | 1-25 | |||
| *** | 98.2 | 86.6 | 86.5 | 91.1 | 90.9 | 84.2 | 90.4 | 93.3 | 2-10 | ||||
| *** | 84.4 | 84.2 | 90.2 | 90.0 | 83.3 | 89.7 | 92.9 | 3-1 | |||||
| *** | 99.5 | 82.5 | 82.7 | 90.6 | 82.5 | 84.8 | 4-4 | ||||||
| *** | 82.3 | 82.5 | 90.9 | 82.2 | 84.6 | 4-3 | |||||||
| *** | 99.8 | 81.0 | 98.3 | 88.4 | 5-351 | ||||||||
| *** | 81.2 | 98.2 | 88.2 | 5-358 | |||||||||
| *** | 80.9 | 83.2 | 5-470 | ||||||||||
| *** | 88.8 | 5-473 | |||||||||||
| *** | Lelystad |
Interpretation von PRRSV-Sequenzen
Die ORF5-Sequenz besteht aus ca. 600 Nukleotiden. Verschiedene Schätzungen deuten darauf hin, dass die Mutationsrate in diesem Gen insgesamt ca. 0,5 % bis 1 % pro Jahr beträgt. Abweichungen der Rate genetischer Veränderungen werden von verschiedenen nicht-viralen Faktoren bestimmt. Das Niveau spezifischer und nicht-spezifischer Immunität gegen PRRSV bei Schweinen hat immense Auswirkungen auf die Virusreplikation und -übertragung, wobei ein immenser inhibitorischer Druck ausgeübt wird, der die Anzahl der Viruskopien verringert. Geringere Viruslasten führen zu niedrigeren Übertragungsraten und überdies zu einer Reduzierung der gesamten Virusreplikation und einer Verringerung der Änderungsrate. Dasselbe Virus kann unter verschiedenen Wirtsbedingungen verschiedene Raten genetischer Veränderung aufweisen. Folglich können wir mitunter höhere oder niedrigere Raten genetischer Veränderung feststellen, die außerhalb des Bereichs von 0,5 % bis 1% pro Jahr liegen.
Die zentrale Frage bei der genetischen Analyse ist, ob zwei Sequenzen nah miteinander verwandt sind (zu zwei Varianten desselben Stamms gehören) oder eigenständig sind (zu zwei Stämmen gehören, die nicht miteinander verwandt sind). Es wird allgemein anerkannt, dass PRRSV-Isolate auf der Grundlage einer prozentualen Ähnlichkeitsgrenze von 97% oder 98 % miteinander verwandt sind oder nicht. Es ist offensichtlich, dass die bloße Berücksichtigung von 2% oder 3% genetischer Unterschiede zwischen zwei Isolaten ohne die Einbeziehung zusätzlicher Kenntnisse zu fehlerhaften Schlussfolgerungen führen kann. Die Unterschiede zwischen den Varianten eines einzelnen Stamms, der einige Jahre lang in einem Bestand zirkuliert, können diesen Grenzwert übersteigen. Die Qualität einer Interpretation des Verwandtschaftsgrads wird durch das Hinzuziehen zusätzlicher Informationen einschließlich der Zeitpunkte und Orte der Isolierung verbessert. Es ist sehr wichtig, neue PRRSV-Sequenzen im Vergleich zu einer breiten Bezugsmenge zu analysieren, die für den Betrieb, das System und die Region repräsentativ ist und ebenso die weltweite genetische Vielfalt wiedergibt.
Die DNA-Sequenzierung von PRRSV-Stämmen kann auf eine enge Verwandtschaft oder Unabhängigkeit der Stämme (Abb. 2) hindeuten, kann aber nicht dazu benutzt werden, den immunologischen Schutz oder PRRS-Ausbrüche in immunen Betrieben vorherzusagen oder zu erklären. Ebenso ermöglicht eine DNA-Sequenzierung nicht die Prognose der klinischen Ergebnisse einer Infektion mit einem bestimmten Stamm, da genetische Marker der Virulenz nicht identifiziert werden.
Derzeit sind überwiegend in Nordamerika, aber auch in Europa viele regionale Projekte zur Bekämpfung oder Eliminierung von PRRS im Gange. Ein umfassendes Bild der genetischen Vielfalt des Virus zu Beginn von Projekten zu seiner Bekämpfung und Eliminierung ist entscheidend für die effektive Überwachung des Verlaufs und der Effektivität der entsprechenden Vorgehensweisen sowie für die Feststellung von Einträgen neuer Viren in Betriebe und die Region.