

Das Influenza-A-Virus (IAV) ist einer der wichtigsten Erreger von Atemwegserkrankungen beim Schwein. Es erhöht die Mortalität und führt zu erheblichen finanziellen Verlusten durch eine geringere Produktivität und die Kosten, die durch die Impfung und die Behandlung entstehen. Außerdem können aufgrund der Anfälligkeit der Schweine für Infektionen mit IAV verschiedener Spezies, insbesondere vom Menschen, neue Viren durch Reassortment auftreten, die sich möglicherweise auch auf die öffentliche Gesundheit auswirken. Folglich kann das Verständnis der genetischen Vielfalt der beim Schwein zirkulierenden Viren dazu beitragen, neue Virusstämme zu identifizieren und Kriterien für die Entwicklung der Interventionsstrategien und deren Verbesserung zur Verfügung zu stellen.
Die genetische Vielfalt des IAV beim Schwein in den USA ist das Ergebnis von Antigendrift und -shift und der Einschleppung des IAV anderer Tierarten in Schweinebestände (Vincent et al., 2008). Im Allgemeinen gibt es die gemeinsame Zirkulation von drei dominanten Subtypen (H1N1, H1N2 und H3N2) und vier genetischen Hauptlinien, die durch die Herkunft der Gensegmente beschrieben werden. Der klassische porzine H1-Virusstamm stammt aus der Pandemie der Spanischen Grippe im Jahr 1918. Der zweite Virusstamm wurde Ende der 1990er Jahre mit dem Hämagglutinin (HA) und der Neuraminidase (NA) nachgewiesen, die vom H3N2-Virus der saisonalen Grippe beim Menschen stammten. Dieser Virusstamm war ein neues reassortiertes Virus, das HA-, NA- und PB1-Gensegmente enthielt, die von der H3N2-Influenza beim Menschen stammten, PB2- und PA-Gensegmente von der Influenza bei Vögeln und NP-, M-, und NS-Gensegmente von der klassischen H1N1-Influenza beim Schwein, weshalb das Virus als „dreifache Reassortante” bezeichnet wurde. Diese Viren reassortierten ihrerseits mit klassischen H1N1-Viren und erwarben dabei das HA und/oder die NA, was zu neuen genetischen Typen von H1N1- und H1N2-Viren führte. Die Konstellation der „dreifach-reassortierten internen Gene” (TRIG) blieb relativ konsistent bezüglich der Ursprünge der Influenzaviren beim Schwein (M-, NP- und NS-Gene), bei den Vögeln (PB2- und PA-Gene) und beim Menschen (PB1). Der dritte Virusstamm resultierte aus wiederholten Infektionen des menschlichen H1-Influenza-A-Virus auf Schweine (bezeichnet als „Spillover“, bei der ein Krankheitserreger einer Spezies auf eine andere übertragen wird) Anfang des 21. Jahrhunderts, wodurch zwei verschiedene Stämme der H1N1- und H1N2-Viren beim Menschen entstanden. Der vierte und neueste Virusstamm entspricht einem „Spillover“ des H3N2-Influenzavirus beim Menschen, das 2010-2011 auftrat. Innerhalb dieser vier HA-Hauptstämme entwickeln sich die Viren weiter und bilden neue genetische Kladen (Abb. 1).

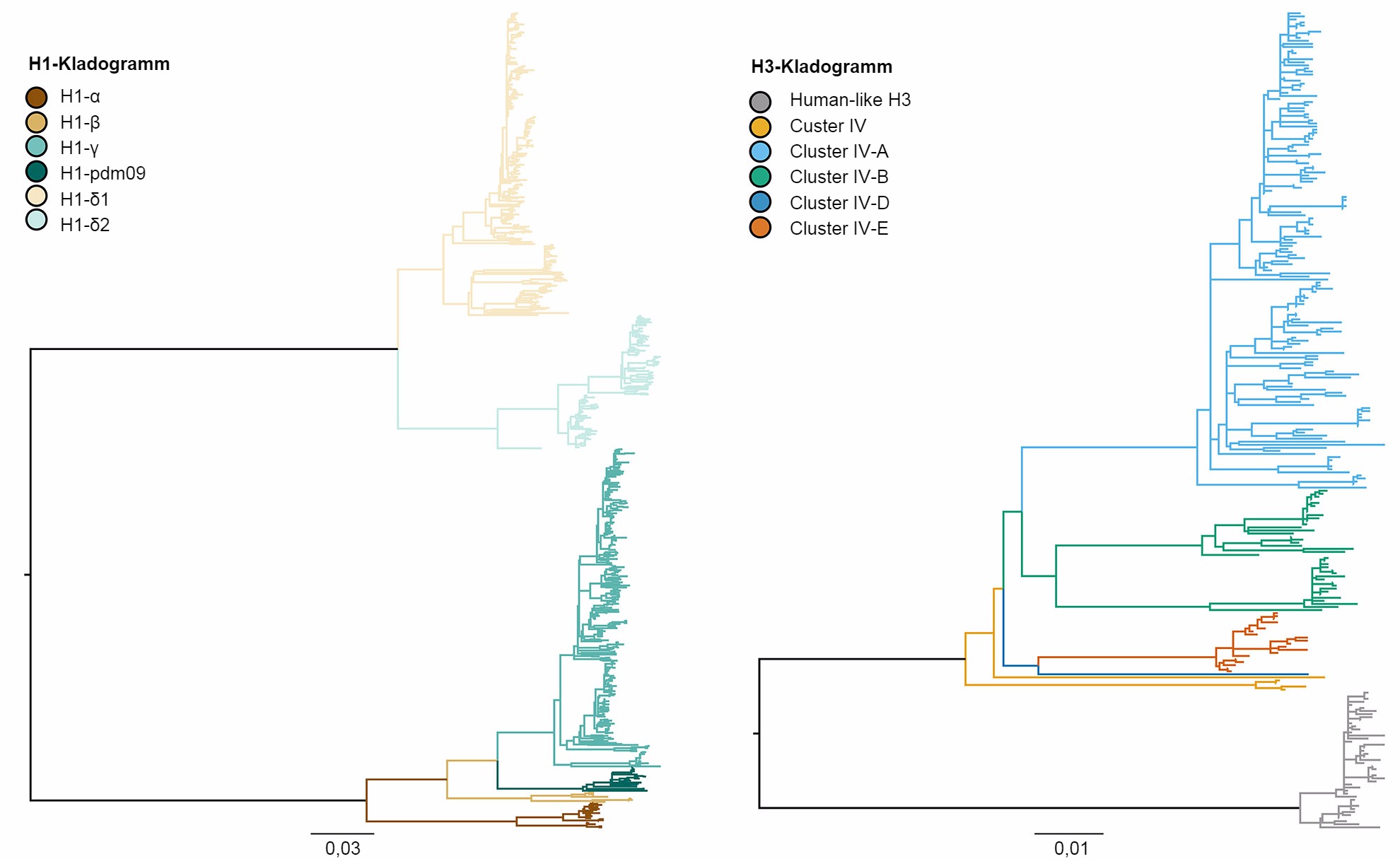
Abbildung 1: Kladogramm, das die genetischen Beziehungen zwischen den Gensequenzen des Hämagglutinins des H1- und des H3-Influenza-A-Virus beim Schwein von 2015 mithilfe der Maximum-Likelihood-Methode beschreibt. Die farbigen Äste kennzeichnen die verschiedenen Kladen. Alle Astlängen sind maßstabsgetreu gezeichnet und der Maßstabsbalken zeigt die Anzahl der ausgetauschten Nukleotide pro Position.
In dem Bemühen, die genetische Vielfalt des Influenza-A-Virus beim Schwein zu verstehen und das 2009 aufgetretene pandemische H1N1-Virus beim Schwein (H1N1pdm09) zu überwachen, startete das Landwirtschaftsministerium der Vereinigten Staaten (USDA) 2009 ein nationales Überwachungssystem, das vom Nationalen Labornetzwerk für Tiergesundheit (NAHLN) umgesetzt wurde. Das System beruht auf anonymen Einsendungen von Erzeugern und Veterinärmedizinern. Die räumlichen Informationen über die Herkunft der Proben sind allerdings nicht sehr detailliert. Erhobene epidemiologische Daten beinhalten den US-Bundesstaat, aus dem sie stammen, den Probentyp, den Grund der Einsendung, das Alter, den Ortstyp, die Testergebnisse und gegebenenfalls wurden die HA-, NA- und M-Gene sequenziert und in der Online-Datenbank für Sequenzen des Nationalen Informationszentrums für Biotechnologie GenBank hinterlegt (Korslund et al., 2012; Anderson et al., 2013). Diese Daten und die weiteren Investitionen in das System boten Einblicke in die Muster der Ausbreitung des IAV, seine genetische Vielfalt während des gesamten Jahres und die Dynamik der Evolution des IAV in Nordamerika seit 2010 bis heute (Anderson et al. 2013; Anderson et al. 2015; Lewis et al. 2014; Rajão et al. 2015).
Die drei Subtypen des Influenzavirus (H1N1, H1N2 und H3N2), die in den Schweinebeständen der USA endemisch sind, wurden seit 2010 bis heute jedes Jahr festgestellt. In den 5 Jahren wurden die H1N1- und H1N2-Subtypen mit ähnlicher Häufigkeit (~35%) festgestellt. Der H3N2-Subtyp repräsentierte während des gesamten Zeitraums ~30% der identifizierten Viren. Zur Beschreibung der genetischen Vielfalt des H1-Virus wurde ein System mit Buchstaben des griechischen Alphabets benutzt, mit dem die Daten der HA-Kladen innerhalb der klassischen Virusstämme (α, β, γ, γ-2 und H1N1pdm09) oder des humanen Virusstamms (δ-1, δ-2) eingeteilt wurden. In ähnlicher Weise teilen sich die Gene des Clusters IV des H3-Stamms in 6 genetische Kladen (A bis F) und eine Klade des human-like H3-Virus auf (Rajão et al. 2015; Kitikoon et al. 2013). Trotz des Potentials von 14 genetischen Kladen zu zirkulieren, war die festgestellte HA-Diversität in US-amerikanischen Schweinebeständen jedoch zu einem großen Teil auf drei Kladen begrenzt. Vom Januar 2015 bis Dezember 2015 gehörten 43% der H1-Isolate zur Klade γ des klassischen Virusstamms und 37% wurden als δ-1 des Virusstamms der saisonbedingten Influenza beim Menschen klassifiziert. Die restlichen 20% der H1-Viren stammten aus den Kladen δ-2, α, H1N1pdm09 und β. Von den 8 potentiellen H3-Kladen repräsentierten die Cluster IV-A die Mehrheit des zirkulierenden H3-IAV beim Schwein in gewerblichen Haltungsbetrieben (61% der H3-Viren im Jahr 2015). Eine wachsende Zahl von Isolaten wurde als H3-Virus des Menschen identifiziert (von 5% im Dezember 2014 bis 25% im Dezember 2015), während die übrigen, sporadisch auftretenden Isolate den Clustern IV-B, IV-C, IV-D, IV-E zugeordnet werden konnten.
Im Rahmen wirksamer Impfprogramme gibt es ein neues Bewusstsein darüber, dass auch die Neuraminidase (NA) aufgrund ihrer zunehmenden Diversität eine wichtige Rolle spielen kann (Sandbulte et al., 2016). Obwohl die Situation dadurch komplizierter wird, besitzt die NA im Vergleich zum HA weit weniger Virusstämme. Die HA ist mit N2-Genen gepaart, die aus einem oder zwei Stämmen der H3N2-Influenza beim Menschen stammen (entweder von 1998 oder von 2002 (Nelson et al., 2011)): N1-Gene des klassischen Virusstamms beim Schwein oder Gene des Virusstamms der saisonalen H1N1-Influenza beim Menschen (Anderson et al., 2013). Bei dem aktuell zirkulierenden IAV gehört das N1 normalerweise zum klassischen Virusstamm (94%) und das N2 zum Virusstamm von 2002 (83%), wobei der Virusstamm von 1998 einen geringen Anteil der IAV-Isolate darstellt, obwohl er regelmäßig nachgewiesen wurde.
Ein zweites Problem bezieht sich auf die räumlichen Muster bei der Diversität des IAV beim Schwein. Auf Grundlage der Veterinärbezirke der Abteilung Tier- und Pflanzenschutzkontrolle des Landwirtschaftsministeriums der Vereinigten Staaten sind die USA in fünf verschiedene Regionen eingeteilt, aus denen über Krankheitsfälle berichtet wird (Abb. 2A). Zwischen den Regionen gibt es feine und wichtige Unterschiede hinsichtlich der Diversität und des Inhalts der Proben, die dem Überwachungssystem des Landwirtschaftsministeriums der Vereinigten Staaten zur Verfügung gestellt werden (Abb. 2B). Während in den Regionen 1 und 2 mehr H1N1 festgestellt wurde, weist Region 3 mehr H1N2 und Region 4 mehr H3N2 auf. Trotz eines Schweinebestands von 1,6 Mio. Schweinen (National Agricultural Statistics Service (NASS) - Landwirtschaftsministerium der Vereinigten Staaten 2012) gibt es sehr wenige Daten aus der Region 5. Die Region 2 weist die größte Diversität hinsichtlich der verschiedenen festgestellten HA- and NA-Kladen auf und die meisten IAV-Isolate vom Schwein stammen aus dieser Region. Insgesamt deutet die Verteilung der HA- and NA-Klade darauf hin, dass möglicherweise regionale oder lokale Entscheidungen bezüglich der Impfstoffkomponenten von Bedeutung sind.
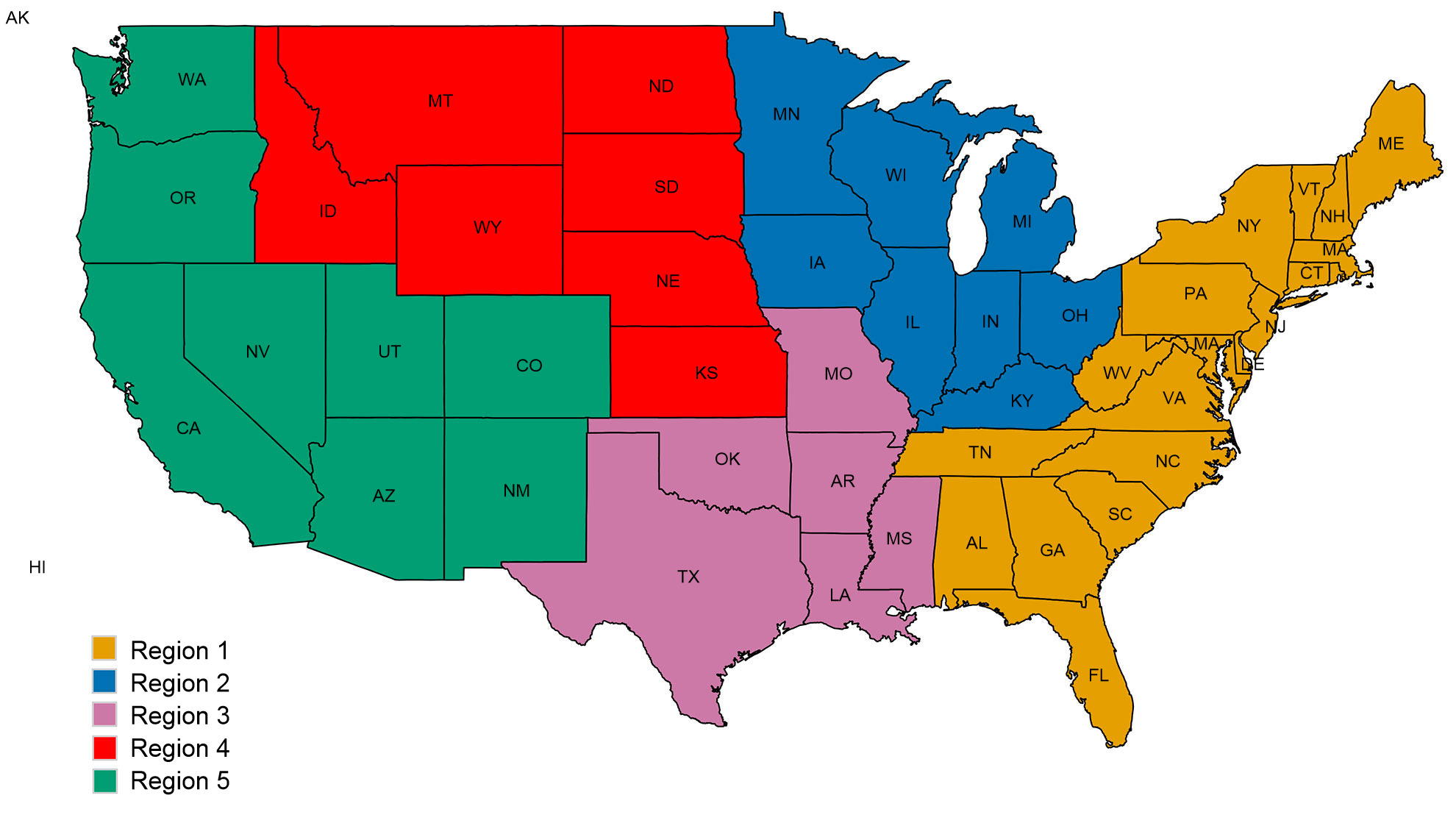
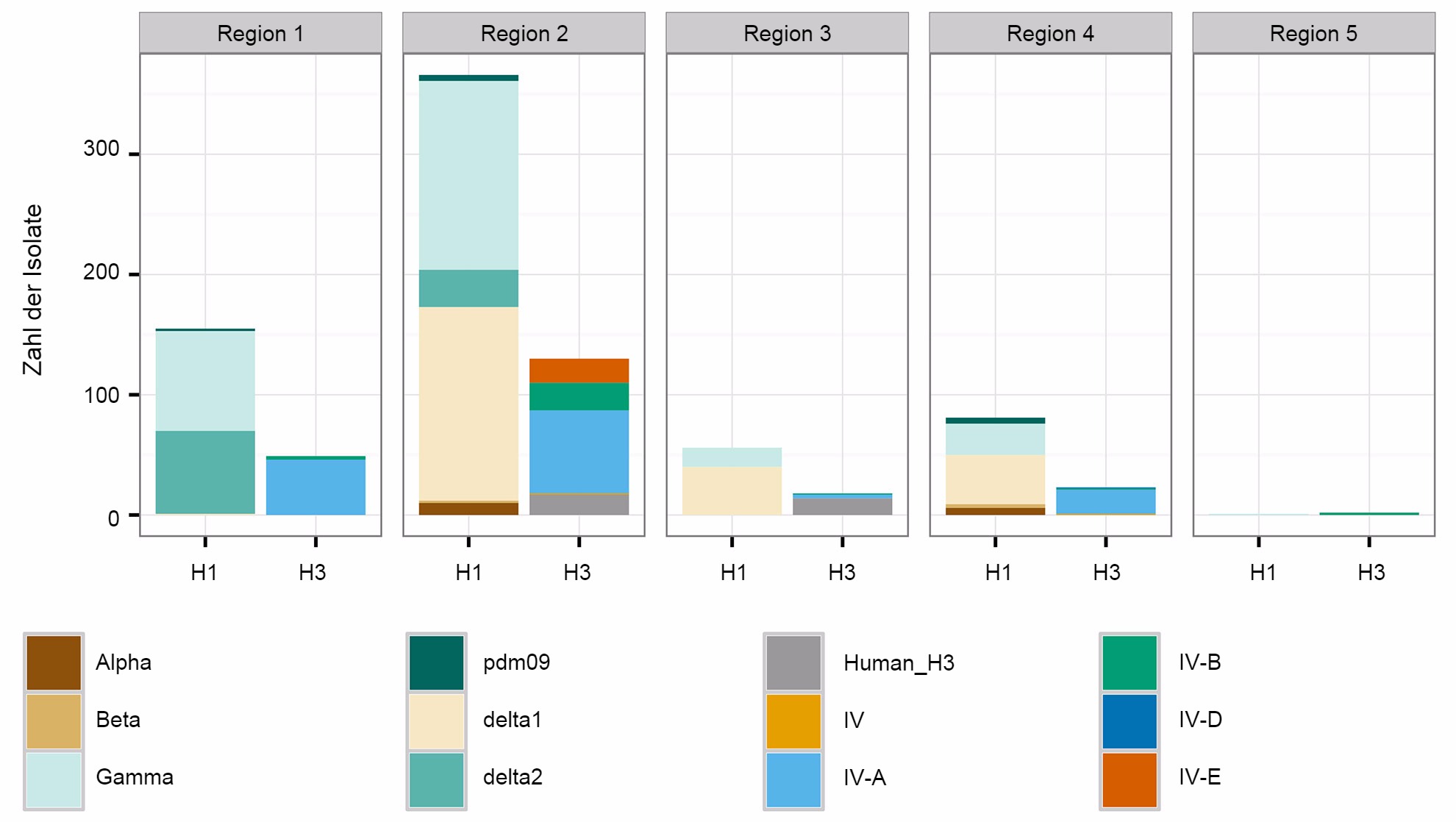
Abbildung 2: Veterinärbezirke der Abteilung Tier- und Pflanzenschutzkontrolle des Landwirtschaftsministeriums der Vereinigten Staaten (USDA-APHIS) (A). Zahl der Schweineinfluenza-A-Isolate, klassifiziert durch phylogenetische Kladen und gemäß Abb. 1. farblich gekennzeichnet, Datenerhebung jeder Region aus dem Jahr 2015 (B)..
Die genetische Vielfalt von IAV ist ein komplexes Thema auf regionaler und insbesondere weltweiter Ebene. In den USA gibt es 17 genetische Kladen, die durch „Spillover“ aus nicht-porzinen (insbesondere humanen) Wirten und den folgenden ökologischen und evolutionsbiologischen Prozessen entstanden und sich halten konnten. Obwohl es verschiedene genetische Kladen gibt, die aus verschiedenen nicht-porzinen „Spillover“ stammen, ist die genetische Vielfalt in den Schweinenbeständen weltweit ähnlich (z. B. Watson et al. 2015; Bahl et al. 2015; Vijaykrishna D et al. 2015). Die Genetik des aktuellen IAV beim Schwein kann zur Durchführung von Studien über die antigenische Evolution und Diversität benutzt werden und diese Untersuchungen sollten mit aktuellen Daten durchgeführt werden, die aus regionalen Beständen stammen. All diese Daten bilden zusammen mit der Einführung geeigneter Impfplattformen die Grundlagendaten zur Auswahl von Impfstämmen und stellen hilfreiche Informationen für die Risikomanagementpolitik zur Förderung und zum Erhalt der landwirtschaftlichen und öffentlichen Gesundheit dar.
